Pdff a Review on Alzheimers Disease Pathophysiology and Its Management an Update
Introduction
Alzheimer'south disease (AD), which was first described past German Bavarian psychiatrist and neurologist Alois Alzheimer in 1907 (i), is the most mutual degenerative fundamental nervous system disease in the elderly. According to the Alzheimer's Association, AD accounts for an estimated 60–80% of dementia cases (2). At nowadays, in that location are 50 million AD patients worldwide, and its incidence doubles every five years after the age of 65 years (3). The main clinical manifestations are cognitive dysfunction, retention loss, and abnormal changes in personality. The pathological cause of Advert is considered to be the senile plaque (SP) formed by amyloid beta (Aβ) and neurofibrillary tangles (NFTs) composed of phosphorylated tau protein, in the hippocampus.
AD can be late onset (LOAD) and sporadic (Lamentable) or early-onset (EOAD) and familial (FAD) (4). FAD is mainly associated with mutations in the Aβ forerunner protein (APP) and presenilin genes PSEN1 and PSEN2, whereas Pitiful has a complex etiology, involving genetic, environmental, metabolic, viral, and other factors (5). Apolipoprotein E (APOE) is a polymorphic protein with iii isoforms, APOE2, APOE3, and APOE4, where APOE4 is the strongest genetic risk factor for Deplorable (6, vii). Co-ordinate to statistics, forty–l% of EOAD and 80% of LOAD are associated with APOE. The strong affinity of APOE for Aβ affects the production, hydrolysis, and emptying of Aβ (8–10). Endogenous expression of APOE4 in stalk-cell-derived neurons promotes the release of phosphorylated tau and predisposes neurons to injury and calcium dysregulation. Interestingly, APOE2 is a protective cistron that reduces the incidence of AD and Aβ aggregating and delays the age of onset (eleven–13). Some researchers have institute that APOE4 expression in mouse models increases oligomer expression and plaque deposition, whereas this is reversed in expression of APOE2 (14). However, there is articulate in vivo bear witness that both APOE2 and APOE4 isoforms are involved in the process of Aβ aggregation and deposition and associated with neurodegeneration, although the result of APOE4 appears to be much stronger than that of APOE2 (15). Regardless, modest-molecule inhibitors of APOE/Aβ interaction may provide a therapeutic option for SAD, which accounts for more than 95% of Ad.
There are many hypotheses to explain AD pathogenesis, involving the amyloid cascade (16), tau hyperphosphorylation (17), neurotransmitters, and oxidative stress (eighteen). Notwithstanding, the underlying causes and optimal treatment plans are still elusive. At present, there are a few drugs available that improve symptoms, by and large targeting Aβ and tau, but these cannot delay progression of the disease. Researchers are beginning to explore new theories of the pathogenesis of AD from different perspectives, such as gamma oscillations, prion transmission, cerebral vasoconstriction, growth hormone secretagogue receptor 1α (GHSR1α)-mediated mechanism, and infection. Discoveries in these areas brand information technology possible to reasonably explain the pathological mechanisms of AD and propose potential effective treatments for Advertising. Herein, we review the two most recognized hypotheses and focus attending on novel developments in the pathophysiology of AD.
Pathogenesis Hypotheses
Most of the recently proposed pathogenic mechanisms are derived from 2 fundamental hypotheses: the amyloid pour hypothesis and the tau hyperphosphorylation hypothesis. Start, nosotros volition review these two accustomed hypotheses and the clinical inquiry targeting them.
Amyloid Cascade Hypothesis
Aβ plaques were first proposed by Paul Blocq and George Mannesco when they discovered "circular accumulation in the brains of elderly patients" in 1892. Later nearly a 100 years of enquiry, Glenner isolated "beta-amyloid" from the meningeal vessels of Alzheimer cases and partially identified the peptide sequence (19). The amyloid hypothesis was start proposed by John Hardy and David Allsop in 1991 (20). Aβ is a transmembrane poly peptide which is produced by hydrolysis of the Aβ precursor protein (APP) via the amyloidogenic pathway. Studies accept shown that APP produces C-terminal fragments nether the hydrolysis of α-, β-, γ-, and η-secretases by three pathways (21) (Figure ane). The first non-amyloidogenic pathological pathway produces products which are neurotrophic and neuroprotective for nervus cells, such as the C-last fragment (CTF)-α, the soluble ectodomain of APP-α (sAPPα), and other smaller fragments, through the involvement of α- and γ-secretases nether normal circumstances. The 2nd pathway is the amyloidogenic pathological pathway in which APP is broken to CTF-β by β-secretase and then different lengths of Aβ peptides past γ-secretase, including Aβ42 which is more than decumbent to aggregation and plaque germination than Aβ40 and has stronger neurotoxicity (22, 23). The third pathway is the culling processing road under physiological atmospheric condition by η-secretase.
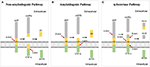
Figure ane. Schematic diagram of the progressive cleavages of the amyloid beta (Aβ) precursor protein (APP) transmembrane domain. Aβ peptide is generated from APP processing via the amyloidogenic pathway (B). (A,C) are non-amyloidogenic pathological pathway nether physiological conditions.
Aβ in SPs is thought to be the initiating cistron in the pathology of Ad (24, 25). Aβ deposited in the hippocampus and basal segment, in the form of neurotoxic amyloid plaques, recruits more than Aβ to form insoluble aggregates and induces mitochondrial damage (26), unstable homeostasis, and synaptic dysfunction (27). Microglia and astrocytes are activated and induce related inflammatory reactions and oxidation. Somewhen, neuronal dysfunction and apoptosis occur, leading to Advertising. Tau protein kinase one tin can be activated by Aβ, leading to abnormal phosphorylation of tau protein and promoting the formation of paired helical filaments (PHFs) and NFT, which accelerate the development of tau pathology (28). Soluble Aβ oligomers are suggested to be more toxic than Aβ cellulose bodies (29). In 2011, Ferreira officially proposed the "Aβ oligomer pathogenic theory," suggesting that soluble Aβ oligomers are the initiating factors leading to a series of pathological changes in Advertizing (30). An increase of Aβ oligomers in the cerebrospinal fluid (CSF) of Advert patients has been reported in many studies (31). Aβ oligomers begin to accumulate in vivo ten years or even decades before clinical symptoms and contribute to long-term potentiation (LTP) inhibition every bit well equally enhanced long-term depression (LTD) (32), past acting on multiple receptors including N-methyl-D-aspartate (NMDA)-type glutamate and α7-nicotinic acetylcholine (α7-nACh) receptors (33), leading to synaptic dysfunction and impaired learning and retentivity (34). Aβ42 oligomers also cause oxidative impairment to synaptic membranes and induce hyperphosphorylation of tau protein (35, 36).
Currently, the goals of therapeutic strategies based on the Aβ hypothesis are to reduce Aβ formation and aggregation and increment Aβ clearance (16, 24, 37) (Table ane). The almost direct action is to reduce Aβ production past controlling BACE1 and γ-secretase activeness (38–40). Nevertheless, γ-secretase inhibitors lack substrate specificity for APP and are toxic to many organs (41). The drug avagacestat, which was the first to undergo clinical trials, has serious side furnishings such as tumors, gastrointestinal reactions, and rashes and did not reach the desired effects; every bit such, related trials have ceased (42–44). The use of Encore'south tarenflurbil, which has good security, has besides been terminated because there is no obvious comeback in cognitive dysfunction (45). Other drugs accept been disappointing to appointment, in some cases performing worse than placebos, with increased adverse reactions (e.g., semagacestat) (44). By dissimilarity, BACE1 inhibitors have higher substrate specificity and are ane of the primary areas for anti-AD drug evolution. However, many Phase III trials have not shown meaning clinical benefits and showed unanticipated adverse side effects (46), such as the inhibitor verubecestat, although it can reduce Aβ in CSF by up to 90% (47, 48). The termination of related clinical trials was announced early in 2018. In July 2019, a Phase II/Three trial of the BACE1 inhibitor umibecestat was also terminated due to cognitive deterioration in participants.

Table one. Part of clinical studies on therapies in Alzheimer's disease (Advertisement).
In addition, animate being experiments have demonstrated that it is feasible to enhance the clearance and degradation of Aβ (49) or promote the delivery of Aβ to the periphery (l). Active immunity and passive immunity are research hot spots. This year, some Phase III trials, such as those for crenezumab and aducanumab, were terminated because they were ineffective and did not attain the primary endpoint, although they were able to reduce Aβ deposition.
Researchers have too attempted to reduce Aβ aggregation to amend brain pathology and noesis in mice (51), past using agents such as the endoglycosylation receptor inhibitor TTP488 (azeliragon) which showed good cognitive improvement in Stage 2 trials (52). Nonetheless, the clinical Phase III trial was terminated considering the desired effect was non achieved.
Recently, researchers at the Academy of Washington developed pocket-sized synthetic alpha-peptides that target and inhibit small toxic oligomers and cake Aβ aggregation at an early stage. Good results were observed in Advert mouse illness models and Caenorhabditis elegans (a nematode model of Advert). However, whether this technique tin exist applied to humans is yet to be studied (53).
Tau Hyperphosphorylation Hypothesis
Tau is a microtubule-associated protein produced by culling splicing of the MAPT gene (54). In 1988, Claude Wischik isolated tau from plaques in the brains of Advertizing patients, demonstrating for the commencement time that tau protein may be the cause of dementia (55). Tau is mainly constitute in neuronal axons of the encephalon (56), combined with microtubules (MTs). The function of tau is to maintain microtubule structure and cytoplasmic ship function (57, 58), maintain synaptic structure and function (59), and regulate neuronal signaling. Tau is likewise a phosphoprotein whose phosphorylation and dephosphorylation may depend on the balance of protein kinase and protein phosphatase activity and is regulated past encephalon development. Under normal conditions, tau has few phosphorylation sites and negatively regulates the bounden of tau to microtubules. Under pathological conditions, the phosphorylation of tau saturated.
The evolution of tau pathology is a complex multifactorial process. Hyperphosphorylated tau in Advertizing patients' brains causes configuration changes and the loss of tubulin polymerization capacity (60, 61), resulting in defective microtubule performance (62).
The elevated levels of cytosolic tau atomic number 82 tau–tau interactions and polymerization to course insoluble PHFs and straight filaments (SFs) that result in the formation of intraneuronal fibrillar deposits known equally NFTs (sixty). NFTs reduce the number of synapses, produce neurotoxicity (63), and cause cell dysfunction (64). Experiments take shown that hyperphosphorylation of tau is positively correlated with the degree of tau aggregation and the pathological severity of AD (65). Tau, rather than Aβ, determines cognitive condition (66). In addition, the acetylation and truncation of tau inhibit its ability to bind to microtubules and also promotes tau assemblage, mitochondrial dysfunction, and synaptic deficits (67–69). Interestingly, p-tau has been shown to spread betwixt cells (lxx). Pocket-sized soluble tau may be more harmful than NFT which tin not only help the spread of pathological tau but can also affect neurodegeneration and cognition (71). In summary, due to its complication, the pathogenesis of pathological tau remains to be elucidated.
In recent years, tau has gained much attention, in part because of the failure of various Aβ-targeting treatments in clinical trials and because tau pathology correlates better with cognitive impairments than do Aβ lesions (Table 1). Inhibitors of kinases and tau aggregation, stabilizers of microtubules, and immunotherapeutic drugs have recently been investigated. Most of them show some toxicity and lack of efficacy, such as the inhibitor of tau aggregation, LMTM (TRx0237) (72). The antifungal molecule epothilone D proved to increase the number of microtubules, reduce the number of abnormal axons, and improve tau-related pathology in a mouse model of tau lesions (73, 74). However, the clinical trial of epothilone D was terminated because of adverse furnishings. AADvac1 as a tau vaccine showed good results in terms of condom and allowed response in Ad patients. Yet, farther research is needed to demonstrate its clinical efficacy (75).
New Insights Into the Pathogenesis of Advertizing
With the prospect of an increasingly aging society, the number of AD patients and sociomedical burdens will increase dramatically. Currently, cholinesterase inhibitors (AChEIs) and the NMDA receptor antagonist are the only therapies for Advertizing (76). However, these tin simply salve symptoms and not delay the progress of AD (77–79). Moreover, three cholinesterase inhibitors, namely, donepezil, rivastigmine, and galantamine, which are canonical by the US Food and Drug Administration, were proven to increase side effects, such as nausea, vomiting, and diarrhea. Although the NMDA receptor adversary memantine showed good effects on improving cognitive function and behavioral disturbance scores (80), it causes severe hypotension, leading to fainting, and falls (81). According to statistics, AD drug development had a high failure rate of 99.6% in the decade between 2002 and 2012 (82). Based on these above unsatisfactory results, researchers are constantly proposing new pathogenic mechanisms.
Gamma Oscillations Ameliorate Pathology and Cognitive Impairment in AD
Gamma oscillations are rhythmic fluctuations of brain waves acquired by activation of local circuits of excitatory and fast-spiking inhibitory neurons in the local field potential (LFP), resonating at twenty–fifty Hz and are associated with numerous college-gild cognitive functions, such as memory germination and attentional pick (83). Changes in gamma oscillations were reported in a variety of neurological diseases including Advertisement. In TgCRND8 transgenic mice, the θ-γ crossover frequency coupling in the hippocampus was dumb earlier plaque germination in the brain (84). Furthermore, the reduction of slow gamma power in CA1 of 3xTg mice tin result in impaired memory function (85). Moreover, hippocampal oscillations were also observed to affect spatial memory in a mouse model of tau pathology (86). These pieces of show suggest that the reactivation of gamma oscillations may play a office in protecting cerebral function in AD.
Surprisingly, studies that replaced endogenous mouse APOE4 with the Advertising-linked human APOE4 gene showed that alleviated gamma impairments after replacement in mice rescued learning and memory deficits (87). Iaccarino and colleagues observed that behaviorally driven gamma oscillations in the Advertisement mouse model are reduced before plaque germination or cognitive turn down begins (88) (Effigy 2). Using optogenetics in the hippocampus of 5XFAD mice and non-invasive light flicker to treat the visual cortex (VC) in multiple mouse models, they institute that 40-Hz gamma oscillation, but not other frequencies, reduced levels of Advertizement pathology. Specifically, Aβ1–40 and Aβ1–42 isoforms in multiple Advertizement mouse models (even in wild-blazon mice), tau phosphorylation staining in the tau P301S mouse model, and fifty-fifty plaque load in anile mice are reduced. Microglial responses were recruited, which is idea to exist a protective function to reduce Aβ past phagocytosis. The verbal manner and consequences of microglial gamma oscillation changes remain to be determined. Nonetheless, these information suggest that reducing neuroinflammation may play an important role in improving neurodegeneration.
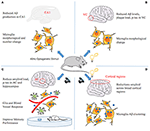
Effigy 2. A series of studies from Li-Huei Tsai indicating that gamma stimulations amend pathology and cognitive impairment in Alzheimer'due south disease (Advert). (A) Gamma induced by optogenetic stimuli reduced amyloid beta (Aβ) production in CA1, increased number and jail cell trunk diameter of microglia, and reduced the process length of microglia compared with the control group, indicating an engulfing state of microglia. The percentage of microglia co-localized with Aβ in the cell body increased, suggesting that gamma stimulation triggers microglia to increase Aβ uptake. (B) Gamma induced by light flicker reduces Aβ levels and plaque load in the visual cortex (VC). Microglia changes similar to (A), except that the number does not alter. (C) In 5xFAD mice, 40-Hz auditory stimulation improves memory performance and reduces amyloid load and tau phosphorylation and seeding in the auditory cortex (Air conditioning) and hippocampus. The changes of microglia like to (A) and the number of astrocytes increased. Furthermore, blood vessel diameter increased. (D) Combined auditory and visual stimulation induces a clustering phenotype response past microglia and reduces amyloid load beyond broad cortical regions.
AD affects multiple brain regions critical to learning and retentivity, such as the hippocampus (HPC) and medial prefrontal cortex (mPFC). Therefore, researchers speculate that gamma oscillators, being generated locally and exhibiting like frequencies in different encephalon regions, can become coupled by anatomical connections between the regions and may be more conducive to alleviating Advertisement pathology and memory loss. Recently, new research by Martorell suggested that auditory tone stimulation has similar effects to visual stimulation in increasing Aβ uptake by microglia, vascular-dilation response, and potential amyloid transvascular transport and in improving spatial and recognition memory (89) (Effigy ii). More importantly, auditory stimulation combined with light-induced gamma oscillations in the hippocampal CA1 and auditory cortex regions of the brain in animal models of AD has unique furnishings: reducing the amyloid load across the cortex, suggesting the possibility of Advertizement-like pathology across larger networks. Long-term handling may exist more than effective than short-term treatment, past transforming neurons into less degraded states, improving synaptic part, enhancing neuroprotective factors, reducing Dna damage in neurons, and reducing the inflammatory response of microglia (90).
However, it is however unclear how not-invasive sensory stimuli are associated with endogenous gamma. Researchers have attempted to use a depression-dose GABAA antagonist on 5XFAD mice and found that the effects of xl-Hz flicker on Aβ levels were completely abrogated, indicating that this process may involve the participation of GABAergic neurotransmission. In improver, whether the benefits can be transformed to humans is also a crucial upshot. Nevertheless, manipulating neural network oscillation disturbances may be a promising strategy to alleviate pathological changes and behavioral deficits associated with neurological diseases.
Aβ and Tau Prions Spread Through the Brains of AD Patients
Prion protein (PrPSc) is a special conformation of a protein encoded by the host, with self-reproduction power, superior infectivity, tenacious viability, and the ability to remain concealed, even surviving in the normal denaturing environment of the digestive organisation. Prions can cause a variety of neurodegenerative diseases in humans, including Creutzfeldt–Jakob affliction (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), and fatal familial insomnia (FFI) in humans (91). These diseases tin occur spontaneously or through genetics or infection.
Studies have shown that Aβ spreads through the brain via a pathogenic conformation similar to PrPSc (92). Brain-derived Aβ and synthetic Aβ from AD patients injected into the brain of transgenic mice showed prion-like appearances (93), which induced plaque formation and extensive degradation of Aβ. Brain extracts from historic period-matched patients without Advertising showed minimal aggregating of Aβ (94). An dissection in a few studies also revealed that some patients had a large amount of Aβ deposition in the encephalon subsequently death, later on receiving dura mater transplantation and cadaveric growth hormone, which may mean that Aβ can be transmitted interpersonally through iatrogenic methods. Prion-like Aβ activity participates in the pathogenesis of Advert. The formation of prion-like Aβ begins in 1 or more encephalon regions and then spreads to other brain regions, reflecting cantankerous-synaptic transmission.
Tau protein is also transmitted in the encephalon in a prion-like manner. Previous studies accept focused solely on total insoluble tau because the affluence of NFT correlates with the extent of brain atrophy and cognitive decline in AD (95). A recent study institute that depression prion-similar tau activity is associated with longer life spans: 100 postmortem brain tissue samples from patients who died of either sporadic or inherited AD showed the presence of both prion-like Aβ and prion-similar tau proteins (96). The activeness of tau prion was inversely proportional to age, which means compared with the Advertising patients with the greatest longevity, patients who died at younger ages due to AD had lower concentrations of both prion-like Aβ and prion-like tau at the time of decease, although NFT increased. This decrease in tau prion-similar activity is concurrent with a decrease in tau phosphorylation, suggesting that biochemical events such as phosphorylation may affect prion-similar tau formation or regulate tau toxicity, although whether the reduction of prion-like tau in elderly Advert patients is due to the conversion of prion-like tau to a more than inert amyloid state, such as total insoluble tau, or due to its reduced product and clearance is still unclear. Therefore, the goal of developing a therapeutic approach of AD for prions remains still to exist accomplished.
Aβ Interact With Hippocampal Ghrelin/GHSR1α Signaling in Advertisement
GHSR1α, a member of the class A G protein-coupled receptor (GPCR) family (likewise known as ghrelin receptor), plays a special role in the hippocampus (97). In the healthy hippocampus, the ghrelin/GHSR1α signal affects the learning, motivational, and hedonic components of eating (98). Moreover, GHSR1α plays a role in hippocampal synaptic physiology and memory maintenance by regulating the dopamine receptor D1 (DRD1) to actuate Ca2+/calmodulin-dependent protein kinase 2 (CaMKII) via the non-canonical Gαq-Ca2+ signaling pathway (97, 99). Hippocampal lesions are i of the earliest lesions to announced in AD and affect cerebral office (100), which may exist related to GHSR1α.
Emerging testify suggests that the loss of GHSR1α induces AD-similar hippocampal synaptic stress and memory deficits (101) (Figure iii). However, the expression of GHSR1α is increased in the hippocampus of AD patients, which may be a compensatory response to the toxic effects of Aβ. About importantly, it is been proven that Aβ combines with GHSR1α, preventing activation of GHSR1α and GHSR1α/DRD1 heterodimerization. The resultant reduced GHSR1α/DRD1 interaction contributes to hippocampal synaptic injury, leading to retention damage (101).
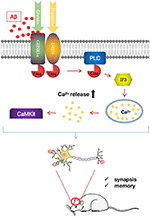
Figure three. Pathways regulating growth hormone secretagogue receptor 1α (GHSR1α)/dopamine receptor D1 (DRD1) interaction past amyloid beta (Aβ) in the hippocampus of patients with Alzheimer's illness (Advertising). Aβ binds directly to GHSR1α and inhibits the activation of GHSR1α and prevented GHSR1α/DRD1 heterodimerization, resulting in synaptic plasticity harm and retention loss. In a mouse model of AD, simultaneous use of the selective GHSR1α agonist MK0677 and the selective DRD1 agonist SKF81297 rescued GHSR1α function from Aβ inhibition, thereby reducing hippocampal synaptic impairment and improving spatial memory.
GHSR1α may exist a target for Advertizement treatment. GHSR1α agonists such as MK0677 and LY444711 showed protective effects in animal and cell models (102, 103). Still, clinical trials of MK0677 in AD patients failed to bear witness clinical benefit. These results may reflect the insensitivity of GHSR1α to activators in Advertising patients. Furthermore, it has been shown that the combined activation of GHSR1α and DRD1 with their selective agonists MK0677 and SKF81297, respectively, rescues hippocampal synaptic office and cognition from Aβ toxicity in young 5XFAD mice (101). Yet, whether MK0677/SKF81297 is benign for older 5XFAD mice requires farther investigation. Yet, this report shows the potential protective effect of this dual agonist intervention in AD.
Still, the roles of GHSR1α may not be limited to the above. Its role in attenuating hippocampal pathology in 5XFAD mice past neurogenesis is not to be ignored (104). Ghrelin has been shown to attenuate hippocampal pathology in 5XFAD mice by neurogenesis. Similarly, coactivation of GHSR1α and DRD1 promotes neurogenesis in the dentate gyrus of 5XFAD mice. GHSR1α dysregulation also has a huge touch on hippocampal metabolic processes and calcium signaling, which is closely related to synaptic activity and hippocampal-dependent memory impairment in elderly and AD patients (105). In addition, GHSR1α tin affect the hypothalamic part and may indirectly drive hippocampal harm (106). In conclusion, the role of GHSR1α in Advertizement cannot be ignored and may provide some promising therapeutic targets.
Aβ Constrict Cognitive Capillaries in Ad Pathology
Cerebrovascular illness can lead to changes in the role of the brain (107). In early AD, angiogenesis damage and cerebral blood period decrease, which are thought to be the first changes of Ad (108). Studies have shown that capillaries contract abnormally in brains of Advertisement patients and that gray blood flow tin can reduce past about 42%. Animal studies have also found that exogenous Aβ tin reduce cerebral blood flow in rats (109), which in turn promotes Aβ production (110).
Recently, a study published in the journal Science demonstrated that in the brains of Advertising patients, Aβ degradation shrank the blood vessels of the brain by most 8.one% and reduced the energy supply, which resulted in a decrease of blood flow of ~50%, which is close to the 42% drib in blood catamenia in the greyness affair of AD patients (111). Aβ involved the generation of reactive oxygen species (ROS), mainly by NOX4 (reduced nicotinamide adenine dinucleotide phosphate oxidase 4), which so triggered the release of endothelin (ET)-1, thereby acting on ETA receptors to evoke pericyte contraction (111). Pericytes became stiff and necrotic after contracting, causing capillary persistent constriction and ischemia (Effigy 4). The Aβ oligomer played an of import role in this process. Experiments conducted on human brain slices fabricated from normal tissue showed that capillaries in encephalon tissues began to contract afterwards exposure to the Aβ composition of oligomers. No similar phenomenon occurred when Aβ monomers were used.
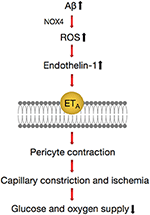
Figure 4. Pathways regulating contractile process of a pericyte around a capillary in the brain. Amyloid beta (Aβ) generates reactive oxygen species (ROS) (via NOX4), which evoke the release of endothelin (ET)-ane, which tin activate wrinkle by binding to ETA. These lead to the release of Ca2+, which evokes contraction of pericytes and brain capillaries, which leads to a decrease of the glucose and oxygen supply to the brain tissue. This pathway can be inhibited by blocking NOX4 with GKT137831 (GKT), blocking ETA receptors with BQ-123, and blocking the ET-evoked contraction by C-blazon natriuretic peptide (CNP).
The vasoconstriction mechanism described suggests that there are some potential therapies for the early handling of AD, such as the NOX4 inhibitor GKT137831 and the vasodilator C-type natriuretic peptide (CNP) (111), which can forestall the wrinkle of blood vessels. This implies that attending should be given to signaling pathways that act directly on neurons and propose novel therapeutic approaches for early intervention in AD past targeting drugs to brain pericytes.
Infection Mechanism in AD
The brain tissue of AD patients exhibits inflammation, such as the activation of complement arrangement factors and microglia (112). The major pathological protein of AD, Aβ, has been shown to exist an antimicrobial peptide, which surrounds invaders in the encephalon and forms plaques to protect the brain from farther damage. Researchers suspected that microbes are involved in the pathogenesis of Advertizing since 1952 (113). Herpes virus is 1 of the potential pathogenic factors of Advertisement. HHV-6A, in particular, is involved in the host regulation of many Advert risk genes such as BACE1 and APBB2 and promotes Aβ precipitation and neuronal loss by inhibiting miR-155 (114). Candida albicans, the pathogen of oral ulcers, was reported to effect in a gelatinous granuloma (FIGG), like to Ad plaques and primary symptoms of suspected AD, such as retentivity loss (115).
Porphyromonas gingivalis, the pathogen of chronic periodontitis (CP), was identified as a risk factor for dementia and AD and caused transient bacteremia through the oral fissure into the claret and then colonized in the organ (116). Gingipain, a cysteine protease consisting of lysine-gingipain (Kgp), arginine-gingipain A (RgpA), and arginine-gingipain B (RgpB), is considered a major virulence cistron of P. gingivalis. P. gingivalis has been identified as an important risk factor for the development of Aβ plaque and AD (Figure 5). A prospective study supported that cognitive function in patients with active CP was significantly reduced inside 6 months compared with AD patients without active CP (117). Infection with P. gingivalis in the oral crenel of mice leads to activation of the complement pathway in the brain (118). Porphyromonas gingivalis lipopolysaccharide has been detected in the human AD brain, suggesting its part in AD (119).
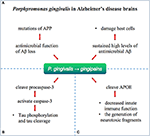
Figure 5. Porphyromonas gingivalis in Alzheimer's disease brains. Porphyromonas gingivalis were identified in the brain of Alzheimer'southward disease patients and chronicle to Aβ, tau, and APOE past gingipains. (A) Loss of biological role of Aβ as an antimicrobial peptide after APP mutation may lead to farther infections. The infection with P. gingivalis results in brain infection of mice and induction of A1-42, which is toxic to host cells. (B) Gingipain proteolysis crusade direct damage or activation of procaspase-3 (which can be broken to activate caspase-3), resulting in Tau phosphorylation and tau cleavage change. (C) APOE4 may be more vulnerable to gingipain slap-up, producing neurotoxic APOE fragments and resulting in decreased innate immune office.
Emerging written report confirmed the pathological role of P. gingivalis and gingipain in Advertizement. The gingipain immunoreactivity (IR) in Advert brain was significantly college than that in not-AD command individuals. Porphyromonas gingivalis is reported to be present in the brain and the CSF of AD patients, indicating that the Deoxyribonucleic acid of P. gingivalis in CSF can exist used equally a differential diagnostic marking. Moreover, gingipain appears in the Advertising brain.
Oral infection with P. gingivalis results in encephalon infection of mice and induction of Aβ1–42, which is toxic to host cells. Loss of biological function of Aβ as an antimicrobial peptide after APP mutation may lead to further infections (120). Tau phosphorylation and tau cleavage are reported to change by direct damage or activation of procaspase-3 (which can be broken to activate caspase-3) by gingipain proteolysis (121). Porphyromonas gingivalis has been hypothesized to relate to human APOE (119), producing neurotoxic APOE fragments and resulting in decreased innate immune function. Effective, selective, permeable brain small molecule gingipain inhibitors are neuroprotective (119). In vivo, it was shown that oral administration of pocket-sized molecule gingipain inhibitors blocked gingipain-induced neurodegeneration and significantly reduced the load of P. gingivalis in mouse brain.
Conclusion
The Aβ cascade hypothesis and tau hyperphosphorylation hypothesis were formulated on the ground of strong genetic, biochemical, and histopathological evidence and were later strengthened by longitudinal biomarker, cognitive, and clinical studies. However, the corresponding clinical drug inquiry has not identified platonic therapeutics. For example, drugs that inhibit Aβ production or accelerate Aβ clearance are highly anticipated. These have probably not yet been identified because whether Aβ accumulation is the crusade, or the result, remains unknown. In the meantime, many researchers have begun to explore the pathological mechanisms of Advertizing from different perspectives. The gamma oscillations caused past auditory and visual stimulation are of note. In addition, there are Aβ and tau prion manual mechanisms, cerebral vasoconstriction, GHSR1α-mediated mechanisms, and infection. All of these offer new strategies for AD research, and based on these, prevention and earlier handling of AD may be possible.
Author Contributions
LF, CM, and XH conceived and wrote the manuscript. YX and CS provided funding. SZ, ZY, ZH, HS, YF, YD, and JY collected resources.
Funding
This work was supported by the National Natural Science Foundation of Mainland china (Grants U190420029, 91849115, and 81530037 to YX; Grants 81771290 and 81974211 to CS; and Grant 81901300 to CM) and National Key R&D Program of China (Grant 2017YFA0105003 to YX).
Disharmonize of Interest
The authors declare that the research was conducted in the absenteeism of any commercial or financial relationships that could be construed equally a potential conflict of interest.
References
three. Brookmeyer R, Gray Due south, Kawas C. Projections of Alzheimer's disease in the United states and the public wellness bear on of delaying disease onset. Am J Public Health. (1998) 88:1337–42. doi: 10.2105/AJPH.88.ix.1337
PubMed Abstruse | CrossRef Full Text | Google Scholar
8. Bales KR, Liu F, Wu Southward, Lin S, Koger D, DeLong C, et al. Man APOE isoform-dependent effects on encephalon beta-amyloid levels in PDAPP transgenic mice. J Neurosci. (2009) 29:6771–9. doi: x.1523/JNEUROSCI.0887-09.2009
PubMed Abstract | CrossRef Total Text | Google Scholar
9. Ye S, Huang Y, Mullendorff Thousand, Dong L, Giedt G, Meng EC, et al. Apolipoprotein. (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure every bit a potential therapeutic target. Proc Natl Acad Sci United states of america. (2005) 102:18700–5. doi: 10.1073/pnas.0508693102
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental furnishings are rescued past pocket-sized-molecule structure correctors. J Biol Chem. (2011) 286:17217–26. doi: 10.1074/jbc.M110.217380
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Serrano-Pozo A, Qian J, Monsell SE, Betensky RA, Hyman BT. APOEepsilon2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. (2015) 77:917–29. doi: 10.1002/ana.24369
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Yamagata Yard, Urakami Yard, Ikeda K, Ji Y, Adachi Y, Arai H, et al. High expression of apolipoprotein E mRNA in the brains with sporadic Alzheimer'south illness. Dement Geriatr Cogn Disord. (2001) 12:57–62. doi: 10.1159/000051236
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Raygani AV, Zahrai M, Raygani AV, Doosti Chiliad, Javadi E, Rezaei M, et al. Association between apolipoprotein E polymorphism and Alzheimer disease in Tehran, Iran. Neurosci Lett. (2005) 375:i–vi. doi: 10.1016/j.neulet.2004.10.073
PubMed Abstract | CrossRef Full Text | Google Scholar
fourteen. Hudry Eastward, Dashkoff J, Roe AD, Takeda South, Koffie RM, Hashimoto T, et al. Gene transfer of homo Apoe isoforms results in differential modulation of amyloid degradation and neurotoxicity in mouse brain. Sci Transl Med. (2013) five:212ra161. doi: 10.1126/scitranslmed.3007000
PubMed Abstract | CrossRef Total Text | Google Scholar
15. Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez Southward, Sullivan PM, et al. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. (2014) 2:75. doi: x.1186/PREACCEPT-1147957959132865
PubMed Abstract | CrossRef Full Text | Google Scholar
xix. Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid poly peptide. Biochem Biophys Res Commun. (1984) 120:885–90. doi: 10.1016/S0006-291X(84)80190-4
PubMed Abstract | CrossRef Total Text | Google Scholar
21. Coronel R, Bernabeu-Zornoza A, Palmer C, Muniz-Moreno M, Zambrano A, Cano E, et al. Role of amyloid precursor protein. (APP) and its derivatives in the biological science and prison cell fate specification of neural stem cells. Mol Neurobiol. (2018) 55:7107–17. doi: 10.1007/s12035-018-0914-ii
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Scheuner D, Eckman C, Jensen M, Vocal Ten, Citron Thou, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo past the presenilin 1 and 2 and APP mutations linked to familial Alzheimer'south affliction. Nat Med. (1996) 2:864–lxx. doi: 10.1038/nm0896-864
PubMed Abstruse | CrossRef Full Text | Google Scholar
23. Di Carlo M, Giacomazza D, San Biagio PL. Alzheimer's disease: biological aspects, therapeutic perspectives and diagnostic tools. J Phys Condens Affair. (2012) 24:244102. doi: 10.1088/0953-8984/24/24/244102
PubMed Abstract | CrossRef Total Text | Google Scholar
26. Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD direct links Abeta to mitochondrial toxicity in Alzheimer's illness. Science. (2004) 304:448–52. doi: 10.1126/science.1091230
PubMed Abstruse | CrossRef Full Text | Google Scholar
28. Vergara C, Houben S, Suain 5, Yilmaz Z, De Decker R, Vanden Dries V, et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced past Alzheimer PHF in vivo. Acta Neuropathol. (2019) 137:397–412. doi: 10.1007/s00401-018-1953-5
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Rasool South, Martinez-Coria H, Wu JW, LaFerla F, Glabe CG. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cerebral function by reducing Aβ degradation and tau pathology in 3xTg-AD mice. J Neurochem. (2013) 126:473–82. doi: 10.1111/jnc.12305
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Ferreira ST, Klein WL. The Aβ oligomer hypothesis for synapse failure and retentiveness loss in Alzheimer's affliction. Neurobiol Learn Mem. (2011) 96:529–43. doi: x.1016/j.nlm.2011.08.003
PubMed Abstract | CrossRef Total Text | Google Scholar
31. Holtta Chiliad, Hansson O, Andreasson U, Hertze J, Minthon L, Nagga Grand, et al. Evaluating amyloid-β oligomers in cerebrospinal fluid as a biomarker for Alzheimer's affliction. PLoS Ane. (2013) eight:e66381. doi: x.1371/journal.pone.0066381
PubMed Abstruse | CrossRef Full Text | Google Scholar
32. Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-β protein dimers isolated directly from Alzheimer'due south brains impair synaptic plasticity and retention. Nat Med. (2008) xiv:837–42. doi: 10.1038/nm1782
PubMed Abstruse | CrossRef Full Text | Google Scholar
34. Li Southward, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. (2009) 62:788–801. doi: 10.1016/j.neuron.2009.05.012
PubMed Abstract | CrossRef Total Text | Google Scholar
35. Butterfield DA, Boyd-Kimball D. Oxidative stress, amyloid-β peptide, and contradistinct primal molecular pathways in the pathogenesis and progression of alzheimer's disease. J Alzheimers Dis. (2018) 62:1345–67. doi: 10.3233/JAD-170543
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer'south illness in APP transgenic mice. Exp Neurol. (2010) 223:422–31. doi: 10.1016/j.expneurol.2009.xi.005
PubMed Abstract | CrossRef Total Text | Google Scholar
37. Karran E, Mercken 1000, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. (2011) 10:698–712. doi: x.1038/nrd3505
PubMed Abstruse | CrossRef Full Text | Google Scholar
38. Chang WP, Koelsch G, Wong Southward, Downs D, Da H, Weerasena V, et al. In vivo inhibition of Abeta product by memapsin ii. (beta-secretase) inhibitors. J Neurochem. (2004) 89:1409–sixteen. doi: ten.1111/j.1471-4159.2004.02452.ten
PubMed Abstract | CrossRef Full Text | Google Scholar
39. McConlogue Fifty, Buttini Thousand, Anderson JP, Brigham EF, Chen KS, Freedman SB, et al. Fractional reduction of BACE1 has dramatic furnishings on Alzheimer plaque and synaptic pathology in APP Transgenic Mice. J Biol Chem. (2007) 282:26326–34. doi: 10.1074/jbc.M611687200
PubMed Abstruse | CrossRef Full Text | Google Scholar
xl. Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and abdominal jail cell differentiation. J Biol Chem. (2004) 279:12876–82. doi: 10.1074/jbc.M311652200
PubMed Abstract | CrossRef Total Text | Google Scholar
41. Geling A, Steiner H, Willem Chiliad, Bally-Cuif L, Haass C. A gamma-secretase inhibitor blocks Notch signaling in vivo and causes a astringent neurogenic phenotype in zebrafish. EMBO Rep. (2002) 3:688–94. doi: 10.1093/embo-reports/kvf124
PubMed Abstruse | CrossRef Full Text | Google Scholar
42. Doerfler P, Shearman MS, Perlmutter RM. Presenilin-dependent gamma-secretase action modulates thymocyte development. Proc Natl Acad Sci USA. (2001) 98:9312–7. doi: ten.1073/pnas.161102498
PubMed Abstruse | CrossRef Full Text | Google Scholar
43. Milano J, McKay J, Dagenais C, Foster-Brownish L, Pognan F, Gadient R, et al. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and consecration of genes known to specify gut secretory lineage differentiation. Toxicol Sci. (2004) 82:341–58. doi: 10.1093/toxsci/kfh254
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Doody RS, Raman R, Farlow Chiliad, Iwatsubo T, Vellas B, Joffe South, et al. A phase iii trial of semagacestat for treatment of Alzheimer'due south affliction. N Engl J Med. (2013) 369:341–fifty. doi: 10.1056/NEJMoa1210951
PubMed Abstract | CrossRef Total Text | Google Scholar
45. Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, et al. Clinical trials and late-stage drug development for Alzheimer's disease: an appraisement from 1984 to 2014. J Intern Med. (2014) 275:251–83. doi: 10.1111/joim.12191
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Identification of beta-secretase. (BACE1) substrates using quantitative proteomics. PLoS Ane. (2009) iv:e8477. doi: 10.1371/periodical.pone.0008477
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, et al. Randomized trial of verubecestat for mild-to-moderate alzheimer's disease. N Engl J Med. (2018) 378:1691–703. doi: 10.1056/NEJMoa1706441
PubMed Abstruse | CrossRef Full Text | Google Scholar
49. Burgos-Ramos Eastward, Hervas-Aguilar A, Aguado-Llera D, Puebla-Jimenez 50, Hernandez-Pinto AM, Barrios V, et al. Somatostatin and Alzheimer's disease. Mol Cell Endocrinol. (2008) 286:104–11. doi: 10.1016/j.mce.2008.01.014
PubMed Abstract | CrossRef Total Text | Google Scholar
l. Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related poly peptide-1: a serial clearance homeostatic mechanism decision-making Alzheimer's amyloid β-peptide elimination from the brain. J Neurochem. (2010) 115:1077–89. doi: 10.1111/j.1471-4159.2010.07002.x
PubMed Abstract | CrossRef Total Text | Google Scholar
51. Hawkes CA, Deng LH, Shaw JE, Nitz M, McLaurin J. Small-scale molecule beta-amyloid inhibitors that stabilize protofibrillar structures in vitro meliorate knowledge and pathology in a mouse model of Alzheimer's disease. Eur J Neurosci. (2010) 31:203–13. doi: x.1111/j.1460-9568.2009.07052.10
PubMed Abstruse | CrossRef Full Text | Google Scholar
52. Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of azeliragon, an oral modest molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of noesis in mild alzheimer'southward disease. J Prev Alzheimers Dis. (2018) 5:149–54. doi: ten.14283/jpad.2018.18
PubMed Abstract | CrossRef Full Text | Google Scholar
53. Shea D, Hsu CC, Bi TM, Paranjapye N, Childers MC, Cochran J, et al. α-Sheet secondary construction in amyloid β-peptide drives aggregation and toxicity in Alzheimer'southward disease. Proc Natl Acad Sci The states. (2019) 116:8895–900. doi: 10.1073/pnas.1820585116
PubMed Abstruse | CrossRef Full Text | Google Scholar
54. Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the homo microtubule-associated poly peptide tau and chromosomal localization of the genes for tau and microtubule-associated poly peptide 2. Brain Res. (1986) 387:271–lxxx. doi: 10.1016/0169-328X(86)90033-1
PubMed Abstruse | CrossRef Full Text | Google Scholar
55. Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci Us. (1988) 85:4884–8. doi: 10.1073/pnas.85.13.4884
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo South, et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B Biol Sci. (2014) 369:20130144. doi: 10.1098/rstb.2013.0144
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan Thou, Wisniewski HM, Binder LI. Aberrant phosphorylation of the microtubule-associated protein tau. (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci Us. (1986) 83:4913–vii. doi: 10.1073/pnas.83.13.4913
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The loftier-affinity HSP90-Chip circuitous recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. (2007) 117:648–58. doi: 10.1172/JCI29715
PubMed Abstract | CrossRef Total Text | Google Scholar
62. Jara C, Aranguiz A, Cerpa W, Tapia-Rojas C, Quintanilla RA. Genetic ablation of tau improves mitochondrial role and cerebral abilities in the hippocampus. Redox Biol. (2018) 18:279–94. doi: x.1016/j.redox.2018.07.010
PubMed Abstruse | CrossRef Full Text | Google Scholar
64. Callahan LM, Vaules WA, Coleman PD. Quantitative decrease in synaptophysin bulletin expression and increase in cathepsin D bulletin expression in Alzheimer disease neurons containing neurofibrillary tangles. J Neuropathol Exp Neurol. (1999) 58:275–87. doi: 10.1097/00005072-199903000-00007
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau poly peptide determines aggregation, synaptic disuse, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. (2008) 28:737–48. doi: 10.1523/JNEUROSCI.2824-07.2008
PubMed Abstract | CrossRef Total Text | Google Scholar
66. Busche MA, Wegmann Due south, Dujardin S, Commins C, Schiantarelli J, Klickstein N, et al. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat Neurosci. (2019) 22:57–64. doi: x.1038/s41593-018-0289-8
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Cotman CW, Poon WW, Rissman RA, Blurton-Jones Yard. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. (2005) 64:104–12. doi: 10.1093/jnen/64.two.104
PubMed Abstract | CrossRef Full Text | Google Scholar
69. Jadhav S, Katina S, Kovac A, Kazmerova Z, Novak M, Zilka N. Truncated tau deregulates synaptic markers in rat model for human tauopathy. Front Cell Neurosci. (2015) ix:24. doi: 10.3389/fncel.2015.00024
PubMed Abstract | CrossRef Full Text | Google Scholar
seventy. Takeda Due south, Wegmann Due south, Cho H, DeVos SL, Commins C, Roe AD, et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer'due south illness encephalon. Nat Commun. (2015) 6:8490. doi: x.1038/ncomms9490
PubMed Abstract | CrossRef Total Text | Google Scholar
71. Shafiei SS, Guerrero-Munoz MJ, Castillo-Carranza DL. Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front Crumbling Neurosci. (2017) 9:83. doi: 10.3389/fnagi.2017.00083
PubMed Abstract | CrossRef Full Text | Google Scholar
72. Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer's affliction: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. (2016) 388:2873–84. doi: x.1016/S0140-6736(16)31275-2
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal Fifty, Liesch J, et al. Epothilones, a new course of microtubule-stabilizing agents with a taxol-similar mechanism of activity. Cancer Res. (1995) 55:2325–33.
PubMed Abstract | Google Scholar
74. Barten DM, Fanara P, Andorfer C, Hoque N, Wong PY, Husted KH, et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J Neurosci. (2012) 32:7137–45. doi: 10.1523/JNEUROSCI.0188-12.2012
PubMed Abstract | CrossRef Total Text | Google Scholar
76. Panza F, Lozupone Grand, Logroscino G, Imbimbo BP. A disquisitional appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. (2019) fifteen:73–88. doi: 10.1038/s41582-018-0116-six
PubMed Abstract | CrossRef Full Text | Google Scholar
78. Aoyagi A, Condello C, Stohr J, Yue W, Rivera BM, Lee JC, et al. Aβ and tau prion-like activities turn down with longevity in the Alzheimer's disease human brain. Sci Transl Med. (2019) xi:eaat8462. doi: 10.1126/scitranslmed.aat8462
PubMed Abstract | CrossRef Full Text | Google Scholar
79. Boada-Rovira M, Brodaty H, Cras P, Baloyannis S, Emre One thousand, Zhang R, et al. Efficacy and safety of donepezil in patients with Alzheimer'due south illness: results of a global, multinational, clinical feel written report. Drugs Aging. (2004) 21:43–53. doi: 10.2165/00002512-200421010-00004
PubMed Abstract | CrossRef Full Text | Google Scholar
80. Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, et al. Randomized trial of verubecestat for prodromal alzheimer's disease. N Engl J Med. (2019) 380:1408–twenty. doi: 10.1056/NEJMoa1812840
PubMed Abstract | CrossRef Full Text | Google Scholar
81. Kishi T, Matsunaga Southward, Oya One thousand, Nomura I, Ikuta T, Iwata North. Memantine for alzheimer's disease: an updated systematic review and meta-analysis. J Alzheimers Dis. (2017) threescore:401–25. doi: 10.3233/JAD-170424
PubMed Abstract | CrossRef Full Text | Google Scholar
82. Gallini A, Sommet A, Montastruc JL. Does memantine induce bradycardia? a report in the french pharmacovigilance database. Pharmacoepidemiol Drug Saf. (2008) 17:877–81. doi: x.1002/pds.1620
PubMed Abstract | CrossRef Full Text | Google Scholar
85. Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot JG, Quirion R, et al. Alterations in hippocampal network oscillations and theta-gamma coupling ascend before Aβ overproduction in a mouse model of Alzheimer's disease. Eur J Neurosci. (2013) 37:1896–902. doi: ten.1111/ejn.12233
PubMed Abstract | CrossRef Full Text | Google Scholar
86. Mably AJ, Gereke BJ, Jones DT, Colgin LL. Impairments in spatial representations and rhythmic coordination of identify cells in the 3xTg mouse model of Alzheimer's disease. Hippocampus. (2017) 27:378–92. doi: 10.1002/hipo.22697
PubMed Abstract | CrossRef Full Text | Google Scholar
87. Booth CA, Witton J, Nowacki J, Tsaneva-Atanasova K, Jones MW, Randall Advert, et al. Altered intrinsic pyramidal neuron properties and pathway-specific synaptic dysfunction underlie aberrant hippocampal network function in a mouse model of tauopathy. J Neurosci. (2016) 36:350–63. doi: 10.1523/JNEUROSCI.2151-15.2016
PubMed Abstract | CrossRef Full Text | Google Scholar
88. Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, et al. Apolipoprotein E4 causes historic period-dependent disruption of tedious gamma oscillations during hippocampal sharp-moving ridge ripples. Neuron. (2016) xc:740–51. doi: 10.1016/j.neuron.2016.04.009
PubMed Abstract | CrossRef Full Text | Google Scholar
89. Iaccarino HF, Singer Air conditioning, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. (2016) 540:230–5. doi: x.1038/nature20587
PubMed Abstract | CrossRef Full Text | Google Scholar
90. Martorell AJ, Paulson AL, Suk HJ, Abdurrob F, Drummond GT, Guan Due west, et al. Multi-sensory gamma stimulation ameliorates alzheimer's-associated pathology and improves cognition. Cell. (2019) 177:256–71.e22. doi: 10.1016/j.prison cell.2019.02.014
PubMed Abstract | CrossRef Full Text | Google Scholar
91. Adaikkan C, Middleton SJ, Marco A, Pao PC, Mathys H, Kim DN, et al. Gamma entrainment binds higher-order encephalon regions and offers neuroprotection. Neuron. (2019) 102:929–43.e8. doi: 10.1016/j.neuron.2019.04.011
PubMed Abstract | CrossRef Full Text | Google Scholar
93. Condello C, Stoehr J. Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer's disease. Neurobiol Dis. (2018) 109(Pt B):191–200. doi: ten.1016/j.nbd.2017.03.014
PubMed Abstract | CrossRef Full Text | Google Scholar
94. Woerman AL, Patel Southward, Kazmi SA, Oehler A, Freyman Y, Espiritu L, et al. Kinetics of man mutant tau prion formation in the brains of 2 transgenic mouse lines. JAMA Neurol. (2017) 74:1464–72. doi: 10.1001/jamaneurol.2017.2822
PubMed Abstract | CrossRef Full Text | Google Scholar
95. Stohr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, et al. Singled-out synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci USA. (2014) 111:10329–34. doi: 10.1073/pnas.1408968111
PubMed Abstruse | CrossRef Total Text | Google Scholar
96. Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. (2013) 12:207–sixteen. doi: x.1016/S1474-4422(12)70291-0
PubMed Abstract | CrossRef Full Text | Google Scholar
97. Kern A, Mavrikaki M, Ullrich C, Albarran-Zeckler R, Brantley AF, Smith RG. Hippocampal dopamine/DRD1 signaling dependent on the ghrelin receptor. Jail cell. (2015) 163:1176–ninety. doi: 10.1016/j.cell.2015.10.062
PubMed Abstract | CrossRef Full Text | Google Scholar
98. Hsu TM, Noble EE, Reiner DJ, Liu CM, Suarez AN, Konanur VR, et al. Hippocampus ghrelin receptor signaling promotes socially-mediated learned food preference. Neuropharmacology. (2018) 131:487–96. doi: 10.1016/j.neuropharm.2017.xi.039
PubMed Abstract | CrossRef Full Text | Google Scholar
99. Seminara RS, Jeet C, Biswas South, Kanwal B, Iftikhar West, Sakibuzzaman M, et al. The neurocognitive effects of ghrelin-induced signaling on the hippocampus: a promising arroyo to alzheimer's disease. Cureus. (2018) 10:e3285. doi: 10.7759/cureus.3285
PubMed Abstruse | CrossRef Full Text | Google Scholar
100. Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive harm. Neurobiol Aging. (2006) 27:1372–84. doi: 10.1016/j.neurobiolaging.2005.09.012
PubMed Abstract | CrossRef Full Text | Google Scholar
101. Tian J, Guo L, Sui S, Driskill C, Phensy A, Wang Q, et al. Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a office in Alzheimer'southward disease. Sci Transl Med. (2019) xi:eaav6278. doi: ten.1126/scitranslmed.aav6278
PubMed Abstract | CrossRef Full Text | Google Scholar
102. Jeong YO, Shin SJ, Park JY, Ku BK, Song JS, Kim JJ, et al. MK-0677, a Ghrelin agonist, alleviates amyloid beta-related pathology in 5XFAD mice, an animal model of alzheimer'due south disease. Int J Mol Sci. (2018) 19:1800. doi: x.3390/ijms19061800
PubMed Abstract | CrossRef Full Text | Google Scholar
103. Eslami M, Sadeghi B, Goshadrou F. Chronic ghrelin administration restores hippocampal long-term potentiation and ameliorates memory impairment in rat model of Alzheimer'south disease. Hippocampus. (2018) 28:724–34. doi: ten.1002/hipo.23002
PubMed Abstract | CrossRef Full Text | Google Scholar
105. Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and retention performance. Nat Neurosci. (2006) ix:381–8. doi: 10.1038/nn1656
PubMed Abstract | CrossRef Full Text | Google Scholar
107. Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, et al. Quantitative imaging of energy expenditure in human brain. Neuroimage. (2012) lx:2107–17. doi: x.1016/j.neuroimage.2012.02.013
PubMed Abstract | CrossRef Full Text | Google Scholar
108. Asllani I, Habeck C, Scarmeas Northward, Borogovac A, Brownish TR, Stern Y. Multivariate and univariate analysis of continuous arterial spin labeling perfusion MRI in Alzheimer's disease. J Cereb Claret Flow Metab. (2008) 28:725–36. doi: x.1038/sj.jcbfm.9600570
PubMed Abstract | CrossRef Total Text | Google Scholar
109. Suo Z, Humphrey J, Kundtz A, Sethi F, Placzek A, Crawford F, et al. Soluble Alzheimers beta-amyloid constricts the cognitive vasculature in vivo. Neurosci Lett. (1998) 257:77–80. doi: x.1016/S0304-3940(98)00814-3
PubMed Abstract | CrossRef Full Text | Google Scholar
110. Sunday X, He G, Qing H, Zhou West, Dobie F, Cai F, et al. Hypoxia facilitates Alzheimer'south disease pathogenesis by upwardly-regulating BACE1 gene expression. Proc Natl Acad Sci U.s.a.. (2006) 103:18727–32. doi: 10.1073/pnas.0606298103
PubMed Abstract | CrossRef Full Text | Google Scholar
111. Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, et al. Amyloid β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science. (2019) 365:eaav9518. doi: 10.1126/science.aav9518
PubMed Abstract | CrossRef Total Text | Google Scholar
112. Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 inflammasome activation of Caspase-ane coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-half dozen activation. Jail cell Decease Differ. (2015) 22:1676–86. doi: 10.1038/cdd.2015.16
PubMed Abstruse | CrossRef Total Text | Google Scholar
113. Sjogren T, Sjogren H, Lindgren AG. Morbus Alzheimer and morbus Selection; a genetic, clinical and patho-anatomical study. Acta Psychiatr Neurol Scand Suppl. (1952) 82:1–152.
PubMed Abstract | Google Scholar
114. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer'southward disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. (2018) 99:56–63.e3. doi: 10.1016/j.neuron.2018.06.030
PubMed Abstract | CrossRef Total Text | Google Scholar
115. Wu Y, Du S, Johnson JL, Tung HY, Landers CT, Liu Y, et al. Microglia and amyloid forerunner poly peptide coordinate control of transient Candida cerebritis with retentiveness deficits. Nat Commun. (2019) 10:58. doi: 10.1038/s41467-018-07991-4
PubMed Abstract | CrossRef Full Text | Google Scholar
116. Kamer AR, Pirraglia E, Tsui W, Rusinek H, Vallabhajosula Southward, Mosconi Fifty, et al. Periodontal affliction assembly with college brain amyloid load in normal elderly. Neurobiol Aging. (2015) 36:627–33. doi: x.1016/j.neurobiolaging.2014.10.038
PubMed Abstract | CrossRef Full Text | Google Scholar
117. Ide Grand, Harris M, Stevens A, Sussams R, Hopkins 5, Culliford D, et al. Periodontitis and cognitive decline in alzheimer's disease. PLoS ONE. (2016) eleven:e0151081. doi: 10.1371/journal.pone.0151081
PubMed Abstract | CrossRef Full Text | Google Scholar
118. Poole Southward, Singhrao SK, Chukkapalli S, Rivera Yard, Velsko I, Kesavalu 50, et al. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J Alzheimers Dis. (2015) 43:67–80. doi: 10.3233/JAD-140315
CrossRef Total Text | Google Scholar
119. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer'south disease brains: evidence for disease causation and treatment with pocket-size-molecule inhibitors. Sci Adv. (2019) v:eaau3333. doi: 10.1126/sciadv.aau3333
PubMed Abstract | CrossRef Full Text | Google Scholar
120. Zhou R, Yang M, Shi Y. Ascendant negative event of the loss-of-function gamma-secretase mutants on the wild-type enzyme through heterooligomerization. Proc Natl Acad Sci Usa. (2017) 114:12731–6. doi: 10.1073/pnas.1713605114
PubMed Abstract | CrossRef Total Text | Google Scholar
121. Urnowey Due south, Ansai T, Bitko V, Nakayama Yard, Takehara T, Barik S. Temporal activation of anti- and pro-apoptotic factors in human gingival fibroblasts infected with the periodontal pathogen, Porphyromonas gingivalis: potential role of bacterial proteases in host signalling. BMC Microbiol. (2006) half dozen:26. doi: x.1186/1471-2180-6-26
PubMed Abstract | CrossRef Full Text | Google Scholar
Source: https://www.frontiersin.org/articles/10.3389/fneur.2019.01312/full
0 Response to "Pdff a Review on Alzheimers Disease Pathophysiology and Its Management an Update"
Enregistrer un commentaire